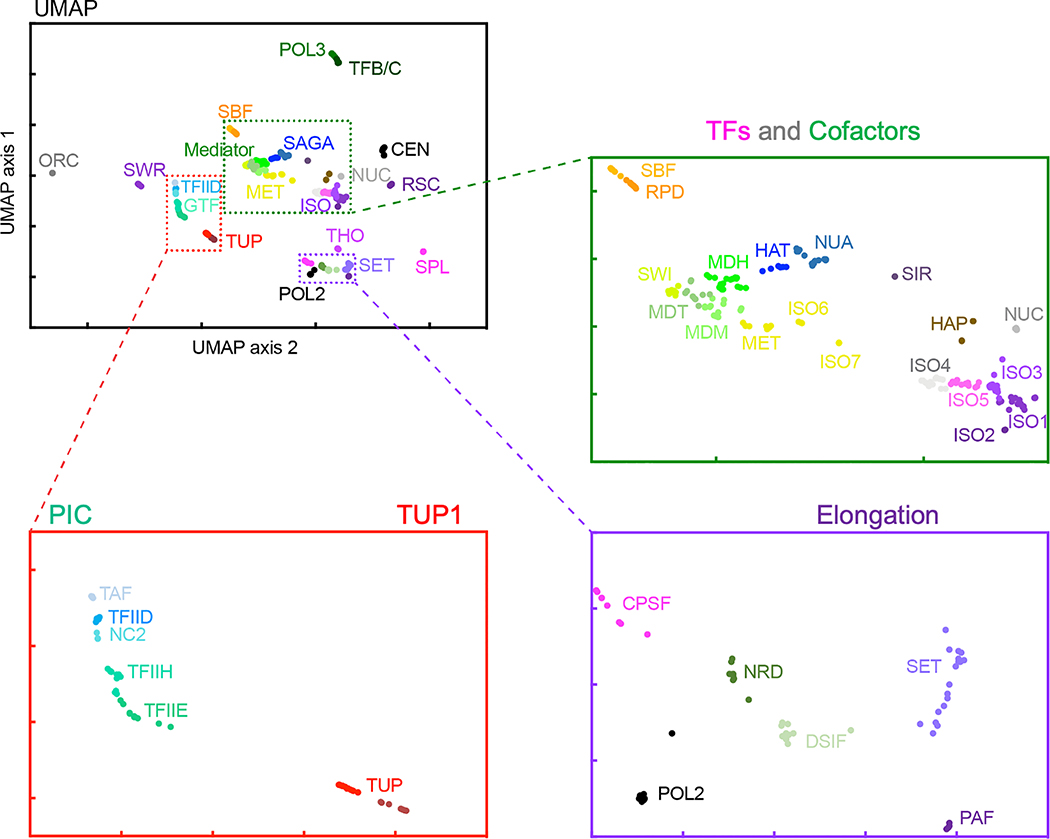
Bioinformatic algorithm development
The development of tools capable of taking advantage of high-resolution data is critical to maximizing the utility of the genomic assays. Our work focuses on integrating diverse datasets across multiple biochemical platforms to derive biologically meaningful insights. Problems we’re interested in include genomic frames of reference (i.e., ArchAlign), high resolution peak-calling (i.e., ChExMix), and deep-learning approaches to understanding the combinatorial nature of cellular regulation (i.e., APSO).
Highlights
-
Adversarial attack of sequence-free enhancer prediction identifies chromatin architecture.
Gafur J, Lang OW, Lai WKM.
Bioinformatics 2025, 41(7). PMID: 40581823; PMCID: PMC12240468. https://doi.org/10.1093/bioinformatics/btaf371 -
Characterizing protein-DNA binding event subtypes in ChIP-exo data.
Yamada N, Lai WKM, Farrell N, Pugh BF, Mahony S.
Bioinformatics 2019, 15;35(6):903-913. doi: 10.1093/bioinformatics/bty703. PMID: 30165373; PMCID: PMC6419906. -
An integrative approach to understanding the combinatorial histone code at functional elements.
Lai WKM, Buck MJ.
Bioinformatics 2013, 29(18):2231-7. doi: 10.1093/bioinformatics/btt382. PMID: 23821650; PMCID: PMC4107033. -
ArchAlign: coordinate-free chromatin alignment reveals novel architectures.
Lai WKM, Buck MJ.
Genome Biology 2010, 11(12):R126. doi: 10.1186/gb-2010-11-12-r126. PMID: 21182771; PMCID: PMC3046486.
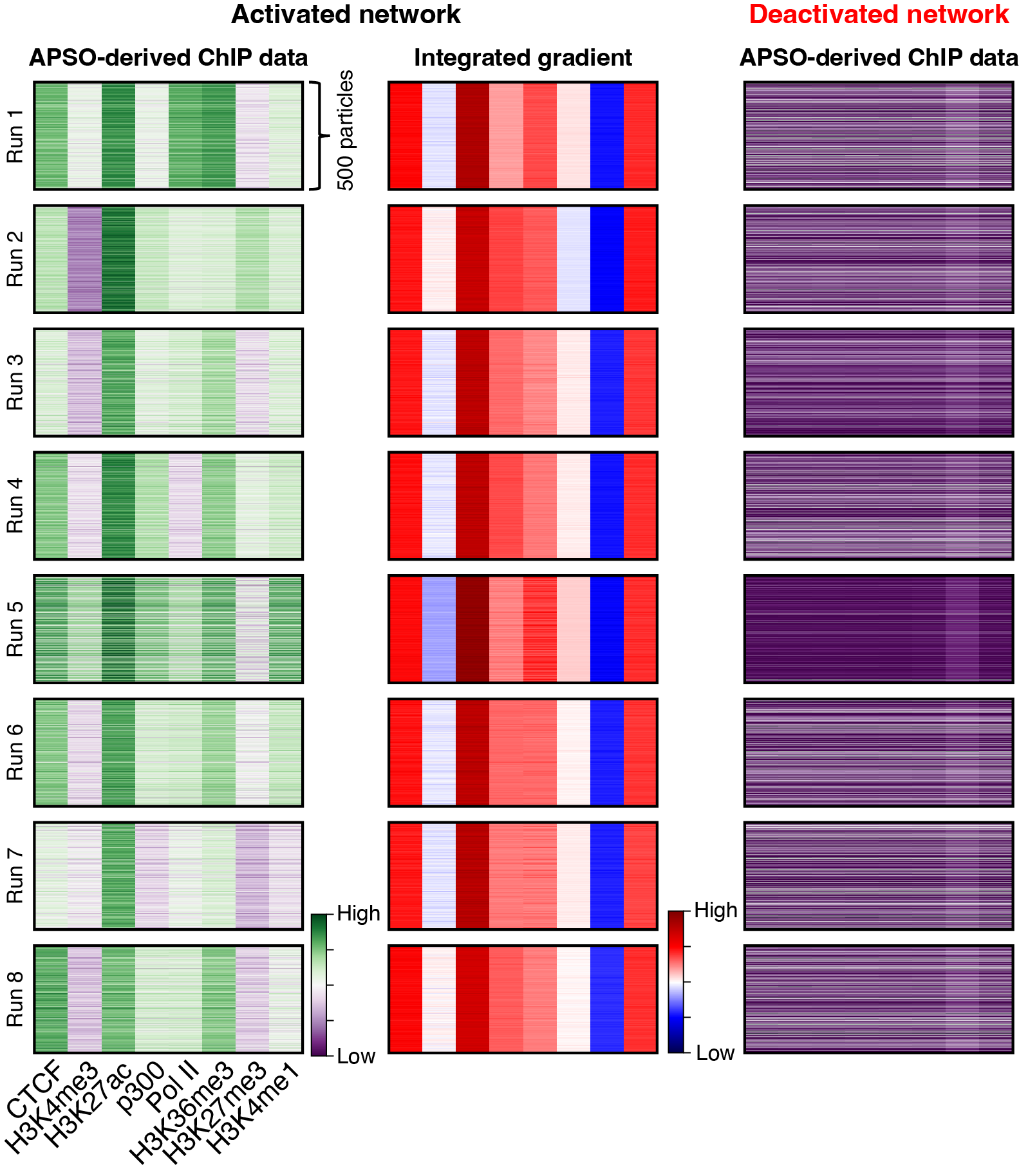
Mechanisms of gene regulation
We apply high-resolution genomic technology to answer basic biological questions. The resolution of our assays provides unprecedented insights into the mechanisms of gene regulation. We use ChIP-exo, PIP-seq, PB-exo, and MNase-ChIP-seq to map the landscape of protein-DNA regulatory mechanisms at base-pair resolution.
Highlights
-
An integrated SAGA and TFIID PIC assembly pathway selective for poised and induced promoters.
Mittal C, Lang O, Lai WKM, Pugh BF
Genome Research 2022, 36(17-18):985-1001. PMID: 36302553 PMCID: PMC9732905 -
Acute stress drives global repression through two independent RNA polymerase II stalling events in Saccharomyces.
Badjatia N, Rossi MJ, Bataille AR, Mittal C, Lai WKM, Pugh BF.
Cell Reports 2021, 34(3):108640. doi: 10.1016/j.celrep.2020.108640. PMID: 33472084; PMCID: PMC7879390. -
Genome-wide determinants of sequence-specific DNA binding of general regulatory factors.
Rossi MJ, Lai WKM, Pugh BF.
Genome Research 2018, 28(4):497-508. doi: 10.1101/gr.229518.117. PMID: 29563167; PMCID: PMC5880240. -
Genome-wide uniformity of human ‘open’ pre-initiation complexes.
Lai WKM, Pugh BF.
Genome Research 2017, 27(1):15-26. doi: 10.1101/gr.210955.116. EPMID: 27927716; PMCID: PMC5204339.
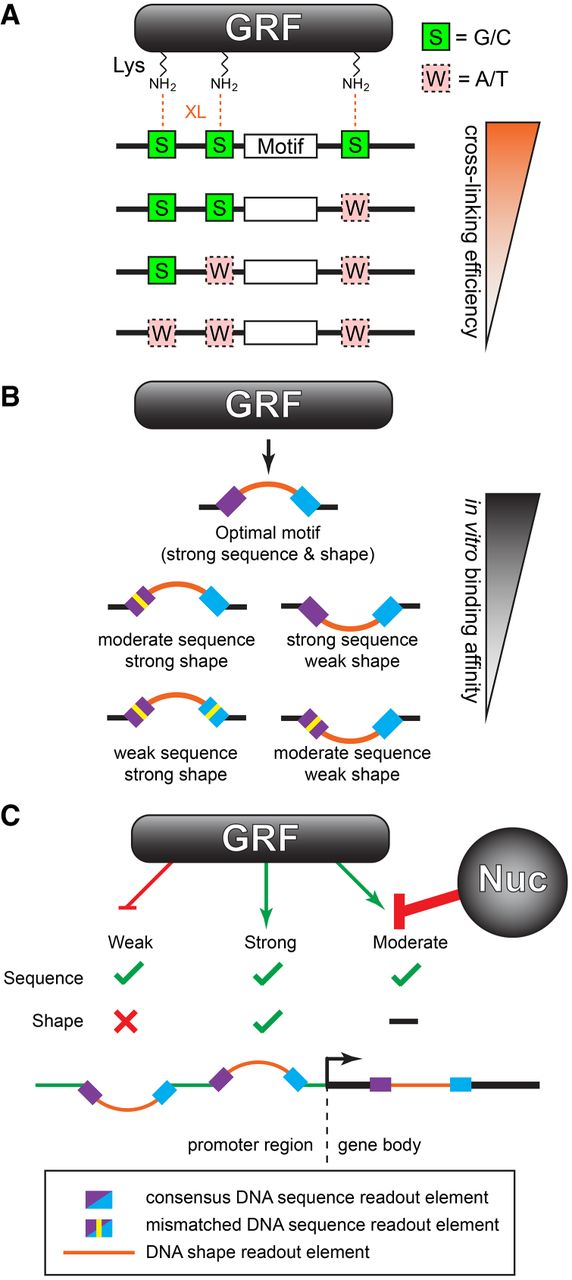
Genomic data generation at scale
Cost and scalability are a huge component of many genomic assays. In collaboration with Pugh lab, we are constantly working to develop significantly cheaper, faster, and higher yield versions of ChIP-exo/seq. We have demonstrated the value of these optimizations, by generating thousands of unique datasets in yeast and human model systems. Our work in the S. cerevisiae system produced the first near-complete (>400 proteins) high-resolution atlas of protein binding. We classified the promoter architecture of every gene in yeast and were able to apply the ChIP-exo assay to identify distinct modes of binding related to gene regulation (i.e., Mediator binding at SAGA genes). We also applied our optimized ChIP-exo assay in collaboration with several other groups according to the NIH’s direct request for us to biochemically validate >1,000 monoclonal antibodies generated through the NIH Common Fund. Of the antibodies tested, 5% produced high-quality data and another 34% produced datasets distinct from background that warrant further investigation. These preliminary epigenomic maps will serve as guides for future hypothesis driven research.
Highlights
-
A high-resolution protein architecture of the budding yeast genome.
Rossi MJ, Kuntala PK, Lai WKM, Yamada N, Badjatia N, Mittal C, Kuzu G, Bocklund K, Farrell NP, Blanda TR, Mairose JD, Basting AV, Mistretta KS, Rocco DJ, Perkinson ES, Kellogg GD, Mahony S, Pugh BF.
Nature 2021, 592(7853):309-314. doi: 10.1038/s41586-021-03314-8. PMID: 33692541; PMCID: PMC8035251. -
A ChIP-exo screen of 887 Protein Capture Reagents Program transcription factor antibodies in human cells.
Lai WKM, Mariani L, Rothschild G, Smith ER, Venters BJ, Blanda TR, Kuntala PK, Bocklund K, Mairose J, Dweikat SN, Mistretta K, Rossi MJ, James D, Anderson JT, Phanor SK, Zhang W, Zhao Z, Shah AP, Novitzky K, McAnarney E, Keogh MC, Shilatifard A, Basu U, Bulyk ML, Pugh BF.
Genome Research 2021, 31(9):1663-1679. doi: 10.1101/gr.275472.121. PMID: 34426512; PMCID: PMC8415381. -
Simplified ChIP-exo assays.
Rossi MJ, Lai WKM, Pugh BF.
Nature Communications 2018, 20;9(1):2842. doi: 10.1038/s41467-018-05265-7. PMID: 30030442; PMCID: PMC6054642.